
Isi
Cystic fibrosis (CF) adalah kelainan bawaan yang mengancam jiwa yang merusak paru-paru dan saluran pencernaan. Ini disebabkan oleh gen yang rusak yang memicu produksi lendir kental yang menyumbat saluran udara dan menghalangi sekresi enzim pencernaan.Gejala bersifat progresif dan seringkali parah, dan mungkin termasuk masalah pernapasan, infeksi paru-paru berulang, pertumbuhan yang buruk, infertilitas pria, dan peradangan kronis pada pankreas, hati, ginjal, dan jantung.
CF dapat didiagnosis dengan tes darah, skrining genetik, dan prosedur yang dikenal sebagai tes klorida keringat.
Meskipun tidak ada obat untuk CF, ada pengobatan yang dapat meningkatkan panjang dan kualitas hidup seseorang.
Ini termasuk teknik pembersihan saluran napas, antibiotik hirup, pengencer lendir, enzim pankreas, diet tinggi kalori, dan obat-obatan generasi baru yang dikenal sebagai modulator CFTR. Dalam kasus yang parah, transplantasi paru-paru mungkin diperlukan.
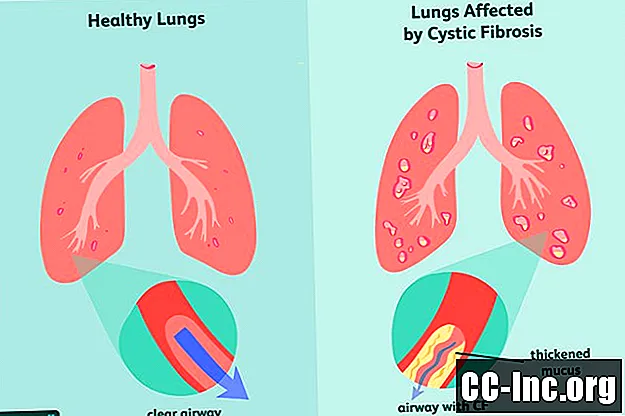
Gejala Fibrosis Kistik
Sebagai kelainan genetik, fibrosis kistik adalah sesuatu yang Anda alami sejak lahir. Ini mungkin atau mungkin tidak hadir dengan gejala pada saat lahir dan seringkali bisa memakan waktu berbulan-bulan atau bahkan bertahun-tahun sebelum tanda-tanda penyakit muncul. Saat itu, paru-paru dan saluran pencernaan mungkin sudah mengalami kerusakan yang tidak bisa diatasi.
Tanda dan gejala awal CF yang paling umum meliputi:
- Penyumbatan tinja pertama bayi (mekonium)
- Kulit terasa asin
- Batuk kronis, mengi atau dahak berwarna
- Kotoran yang kendur, berminyak, dan biasanya berbau tidak sedap
- Infeksi paru-paru, seringkali berulang
- Pertumbuhan yang buruk dan kegagalan untuk berkembang
Kecuali gejala ini dapat dikontrol, tekanan pada paru-paru (dan ketidakmampuan untuk menambah berat badan) dapat memiliki efek kumulatif, mempengaruhi banyak organ dan meningkatkan risiko komplikasi penyakit.
Beberapa komplikasi yang lebih khas meliputi:
- Pubertas tertunda
- Bronkiektasis (penebalan kronis dinding paru-paru)
- Penurunan berat badan
- Pankreatitis (radang pankreas)
- Infertilitas pria
- Hipertensi pulmonal (tekanan darah tinggi di paru-paru)
- Batu empedu
- Diabetes terkait fibrosis kistik
- Cor pulmonale (gagal jantung sisi kanan)
- Sirosis (jaringan parut fungsional hati)
Karena CF menyebabkan cedera progresif pada sel dan jaringan, kerusakan apa pun yang terjadi pada paru-paru dan organ lain sebagian besar tidak dapat disembuhkan. Kematian paling sering terjadi akibat gagal napas, diikuti oleh gagal jantung dan gagal hati.
Gejala Fibrosis Kistik
Penyebab
Cystic fibrosis disebabkan oleh mutasi gen cystic fibrosis transmembrane receptor (CFTR), yang bertanggung jawab untuk memproduksi protein CFTR. Ini adalah protein yang dibutuhkan tubuh untuk mengatur aliran garam dan air masuk dan keluar sel. . Jika protein berubah bentuk atau cacat, dapat menyebabkan dehidrasi pada permukaan sel, yang menyebabkan penebalan lendir di sekitarnya.
CF adalah gangguan resesif autosomal, yang berarti Anda harus mewarisi mutasi CFTR dari ibu dan ayah Anda untuk mengidap penyakit tersebut. Jika Anda hanya mewarisi satu gen yang rusak, Anda tidak akan menderita CF tetapi malah menjadi pembawa gen yang bermutasi.
Anda dapat mewarisi penyakit ini jika masing-masing orang tua Anda memiliki mutasi CFTR atau CF itu sendiri. Jika kedua orang tua adalah pembawa, Anda akan memiliki:
- 25 persen kemungkinan memiliki CF.
- 50 persen kemungkinan menjadi pembawa
- 25 persen kemungkinan tidak terpengaruh
Di sisi lain, jika salah satu orang tua Anda adalah pembawa dan yang lainnya memiliki CF, Anda memiliki peluang 50/50 untuk memiliki CF atau menjadi pembawa.
Cystic fibrosis adalah salah satu penyakit genetik yang lebih umum, mempengaruhi kira-kira satu dari setiap 2.500 bayi yang lahir di Amerika Serikat.
Ini paling umum di antara Kaukasia dan Hispanik, dan lebih jarang terjadi pada orang keturunan Afrika atau Asia.
Faktor Risiko Fibrosis KistikDiagnosa
Ada beberapa tes yang digunakan untuk mendiagnosis fibrosis kistik. Mereka bekerja baik dengan secara langsung mendeteksi mutasi CFTR atau secara tidak langsung mengukur perubahan biologis yang konsisten dengan penyakit. Metode diagnosis dapat bervariasi selama kehamilan, saat bayi lahir, atau kapan saja setelahnya.
Panduan Diskusi Dokter Cystic Fibrosis
Dapatkan panduan cetak kami untuk janji dengan dokter Anda berikutnya untuk membantu Anda mengajukan pertanyaan yang tepat.

Dari dua tes standar yang biasa digunakan untuk mendiagnosis CF:
- Pengujian klorida keringat, juga dikenal sebagai tes keringat, mengukur jumlah klorida pada kulit. Karena CF mengganggu transfer garam ke dan dari sel, maka akan terjadi penumpukan garam di keringat.
- Pengujian CFTR genetik digunakan untuk mendeteksi mutasi mutasi CFTR yang paling umum. Meskipun ada lebih dari 2.000 mutasi CFTR yang diketahui menyebabkan fibrosis kistik, 23 yang termasuk dalam panel standar mewakili tersangka yang paling mungkin.
Selama kehamilan, tes genetik CFTR dapat digunakan untuk menguji cairan yang diperoleh melalui amniosentesis atau sel yang diekstraksi melalui chorionic villus sampling (CVS).
Skrining bayi baru lahir juga digunakan secara standar untuk mendiagnosis CF dan saat ini diamanatkan di semua 50 negara bagian dan District of Columbia. Apa yang diperlukan akan berbeda-beda tergantung di Amerika Serikat tempat Anda tinggal. Jika hasil skrining bayi baru lahir positif, tes keringat akan digunakan untuk memastikan diagnosis.
Bagaimana Fibrosis Kistik DidiagnosisPengobatan
Meskipun tidak ada obat untuk fibrosis kistik, kemajuan dalam pengobatan telah memperpanjang umur mereka yang hidup dengan penyakit tersebut.
Tujuan pengobatan CF ada empat: untuk mencegah infeksi, mempertahankan fungsi paru-paru, menormalkan pencernaan, dan memperlambat perkembangan penyakit.
Di antara alat terapeutik yang digunakan untuk menangani CF:
- Teknik pembersihan jalan napas (ACTs) dilakukan untuk mengeluarkan dan mengeluarkan lendir yang terkumpul dari paru-paru. Tekniknya termasuk batuk tergesa-gesa, perkusi dada, atau osilasi dinding dada.
- Diet tinggi lemak dan kalori tinggi digunakan untuk mengkompensasi malabsorpsi lemak, protein, dan nutrisi di usus.
- Suplemen enzim pankreas digunakan untuk meningkatkan enzim pencernaan yang tidak dapat diproduksi oleh pankreas karena penumpukan lendir yang berlebihan.
- Antibiotik diminum setiap hari untuk mencegah infeksi paru-paru bakteri.
- Mucolytics-Obat yang digunakan untuk mengencerkan lendir sebelum ACTs-dapat digunakan.
- Modulator CFTR adalah kelas obat baru yang dapat memperbaiki kerusakan tertentu pada protein CFTR dan mengembalikan fungsi pengaturannya.
- Terapi oksigen dapat digunakan selama episode akut ketika pernapasan Anda sangat terganggu.
- Nutrisi enteral, juga dikenal sebagai pemberian makan tabung, dapat digunakan jika Anda tidak dapat mempertahankan berat badan melalui nutrisi normal.
- Transplantasi paru-paru dipertimbangkan ketika paru-paru Anda tidak dapat lagi mendukung kelangsungan hidup tanpa ventilasi mekanis.
Mengatasi
Pada tahun 1938, ketika cystic fibrosis pertama kali diklasifikasikan sebagai penyakit, anak-anak jarang hidup setelah tahun pertama kehidupan mereka. Pada 1980-an, orang bisa berharap untuk hidup selama 20 sampai 25 tahun. Saat ini, gambaran tersebut telah berubah sepenuhnya dengan orang yang hidup sehat hingga usia 40-an dan bahkan 50-an jika pengobatan dimulai sejak dini dan ditaati.
Ini tidak berarti bahwa CF tidak seserius sebelumnya. Ini adalah peristiwa yang mengubah hidup, membutuhkan ketekunan dan konsistensi untuk tidak hanya mengatasi penyakit tetapi juga menjalani standar hidup setinggi mungkin.
Untuk itu, Anda perlu menormalkan CF dalam hidup Anda dengan menetapkan rutinitas dan praktik untuk menghindari pasang surut yang dapat menyebabkan stres dan meningkatkan kecacatan. Di antara pertimbangan tersebut, Anda perlu:
- Kelola nutrisi Anda. Orang dengan CF sering membutuhkan kalori dua kali sehari daripada orang lain.
- Berolahragalah secara teratur. Rutinitas kebugaran idealnya melibatkan minimal 20 hingga 30 menit aktivitas aerobik tiga kali seminggu. Temukan sesuatu yang menyenangkan yang dapat Anda lakukan seumur hidup.
- Tetap terhidrasi dengan baik. Melakukannya membuat paru-paru dan usus bekerja dengan baik. Bergantung pada usia Anda, Anda harus minum tidak kurang dari enam hingga delapan gelas air per hari.
- Lakukan pembersihan jalan napas dengan benar. Saat kesehatan Anda perlu berubah, demikian juga jenis alat izin yang Anda butuhkan. Bicaralah dengan ahli paru atau ahli terapi fisik Anda jika Anda tidak mencapai hasil yang seharusnya.
- Cari dukungan. Selain teman dan keluarga, Anda dapat menghubungi cabang Cystic Fibrosis Foundation (CFF) terdekat untuk terhubung ke jaringan dukungan di wilayah Anda.
- Cari bantuan keuangan. CFF menawarkan layanan yang membantu keluarga mengatasi biaya pengobatan CF yang tinggi dengan lebih baik.
Sebuah Kata Dari Sangat Baik
Meskipun pemeriksaan bayi baru lahir telah secara dramatis meningkatkan angka diagnosis CF pada bayi, lebih dari 25 persen diagnosis hanya dibuat selama masa kanak-kanak, remaja, dan dewasa awal.
Ini bermasalah karena diagnosis dan pengobatan dini dapat mencegah banyak komplikasi CF yang lebih parah sebelum kerusakan serius dapat dilakukan. Meskipun pengobatan tidak dapat menghentikan atau membalikkan penyakit, ini dapat memastikan lebih banyak tahun bebas penyakit.
Untuk tujuan ini, penting untuk mengetahui gejala awal CF dan berbicara dengan dokter Anda jika Anda mencurigai anak Anda mungkin mengidap penyakit tersebut. Hal ini terutama berlaku di negara bagian yang melakukan skrining hanya dengan tes darah IRT, yang dapat mengakibatkan sebanyak 5 persen anak-anak mengalami diagnosis tertunda atau hasil negatif palsu, menurut penelitian dari Fakultas Kedokteran dan Kesehatan Masyarakat Universitas Wisconsin. .
Gejala Apa Yang Dapat Anda Harapkan Dengan Cystic Fibrosis?